Assistant Professor University of Miami Leonard M. Miller School of Medicine Miami, Florida, United States
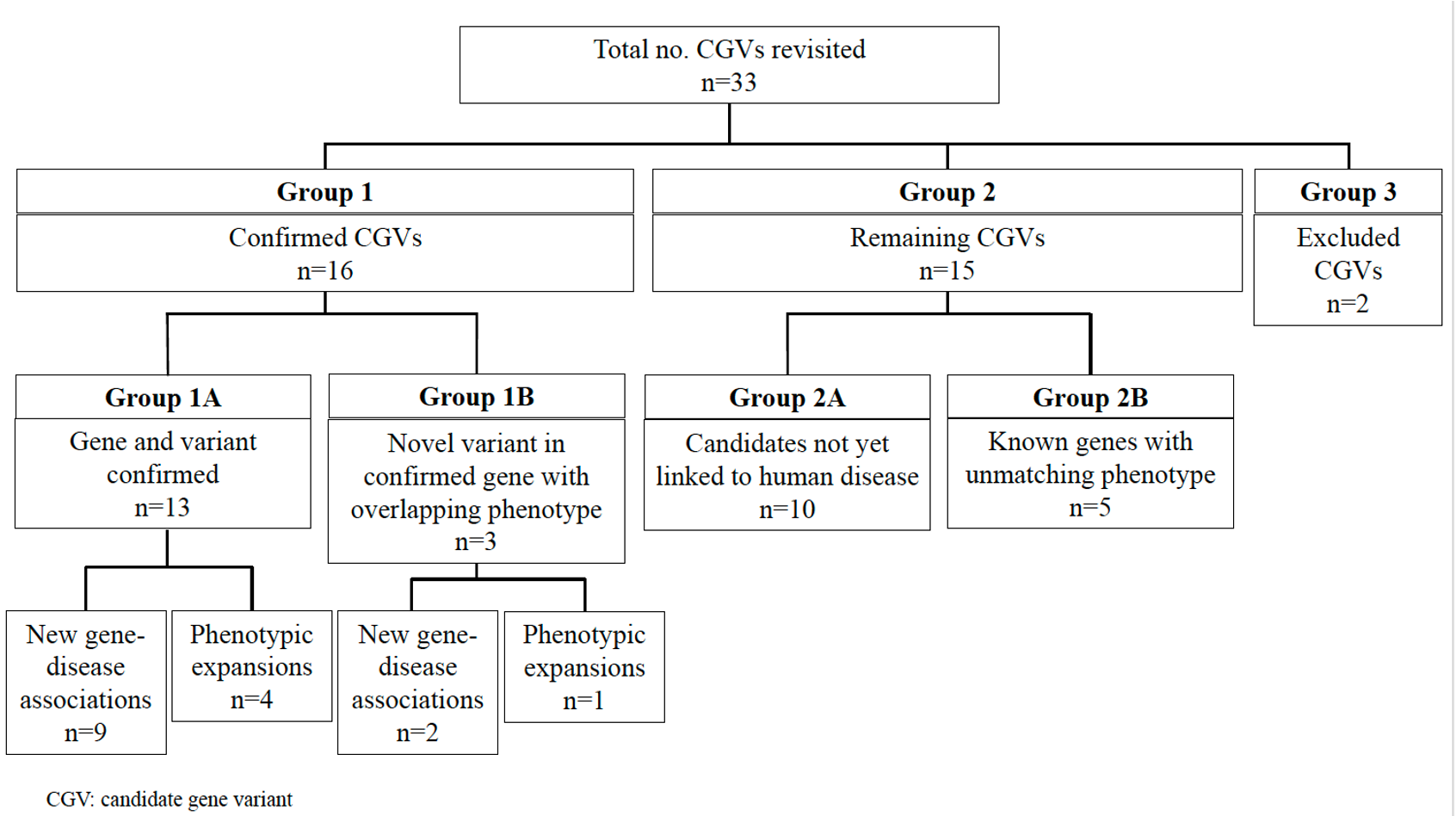
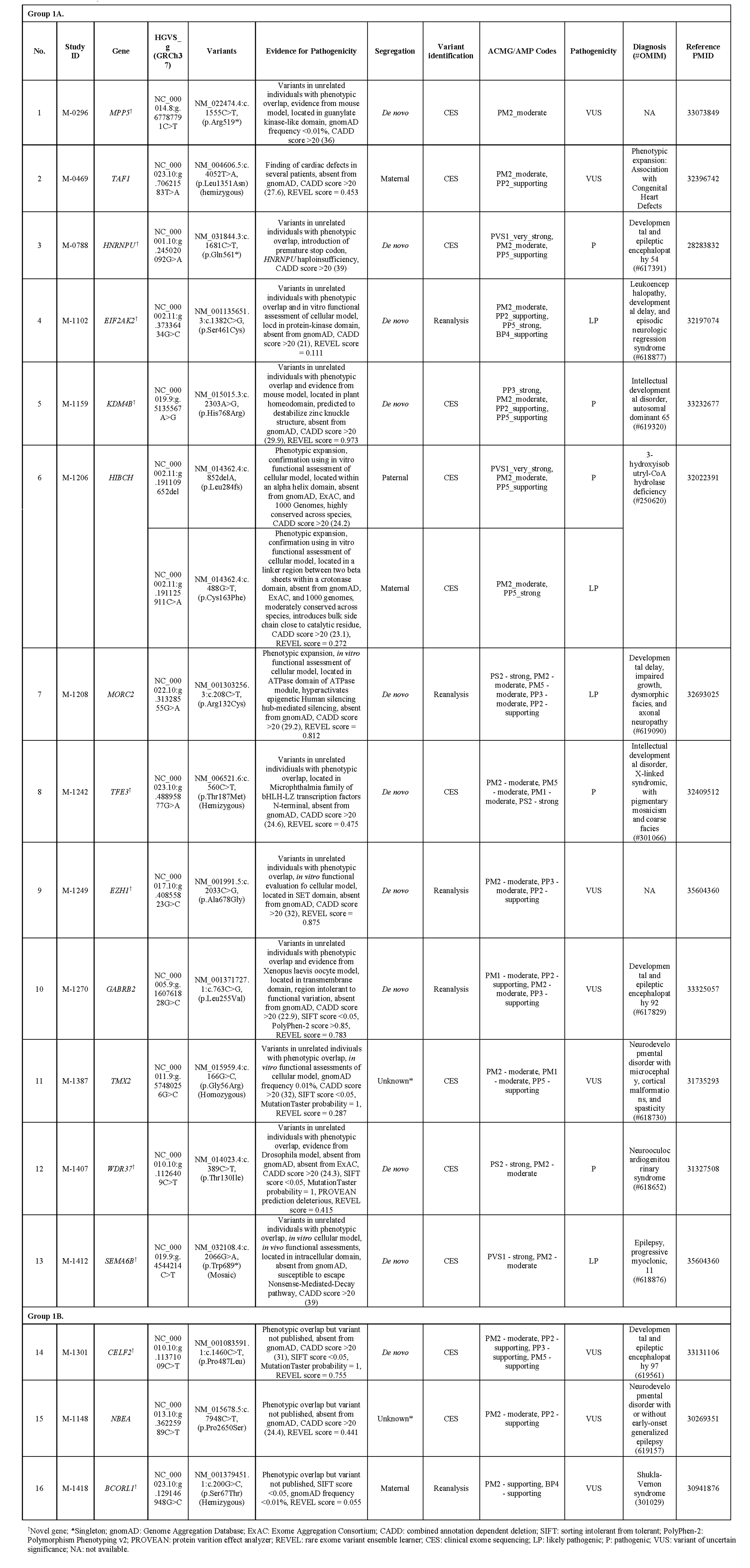
Background: Advances in bioinformatic tools, paired with the ongoing accumulation of genetic knowledge and periodic reanalysis of genomic sequencing data, have led to an improvement in genetic diagnostic rates. Candidate gene variants (CGVs) identified during sequencing or reanalysis but not yet implicated in human disease or associated with a phenotypically distinct condition are often not revisited, leading to missed diagnostic opportunities. Objective: In a previous pilot study, we reanalyzed 102 undiagnosed cases following clinical exome sequencing and were able to establish new genetic diagnoses in six individuals and identify 33 cases with CGVs. To further analyze these CGVs, we revisited those 33 cases to identify the disease-causing variants. Design/Methods: Participants’ clinical records were reviewed to obtain the most up-to-date information regarding genetic diagnoses. The CGVs were evaluated via literature review and by discussion with the Manton Center Gene Discovery Core team consisting of genetic counselors, bioinformaticians, researchers, and both internal and referring clinicians. The NCBI PubMed database and the Human Gene Mutation Database were queried for each CGV. Overall, the CGVs were classified into three groups after reevaluation: Confirmed (Group 1), Candidate (Group 2), and Excluded (Group 3). Within Group 1, CGVs were subdivided: published variants in confirmed disease genes, including both new gene-disease associations and phenotypic expansions (Group 1A) and novel variants in published disease-associated genes with clinical presentations matching the published patient phenotype (Group 1B). Group 2 was subdivided into two categories as well: variants in genes that remain unlinked with human disease (Group 2A) and those that are now linked to human disease, but our patient's phenotype does not overlap with the published phenotype (Group 2B). Results: We revisited those 33 cases and established that 16 of these CGVs now have published disease-associations or phenotypic expansions consistent with our patient’s clinical presentations (49%). Of the remaining 17 CGVs, 15 (45%) are still candidates, and 2 (6%) have been ruled out as disease-causing. Our experience demonstrates that CGV identification and ensuing extensive work up are critical towards disease gene discovery and ending diagnostic odysseys.
Conclusion(s): These results emphasize the need to focus on previously identified CGVs during sequencing or reanalysis and the importance of sharing that information with researchers around the world, including relevant functional analysis to establish disease causality.