Research assistant professor University of Miami Leonard M. Miller School of Medicine Miami, Florida, United States
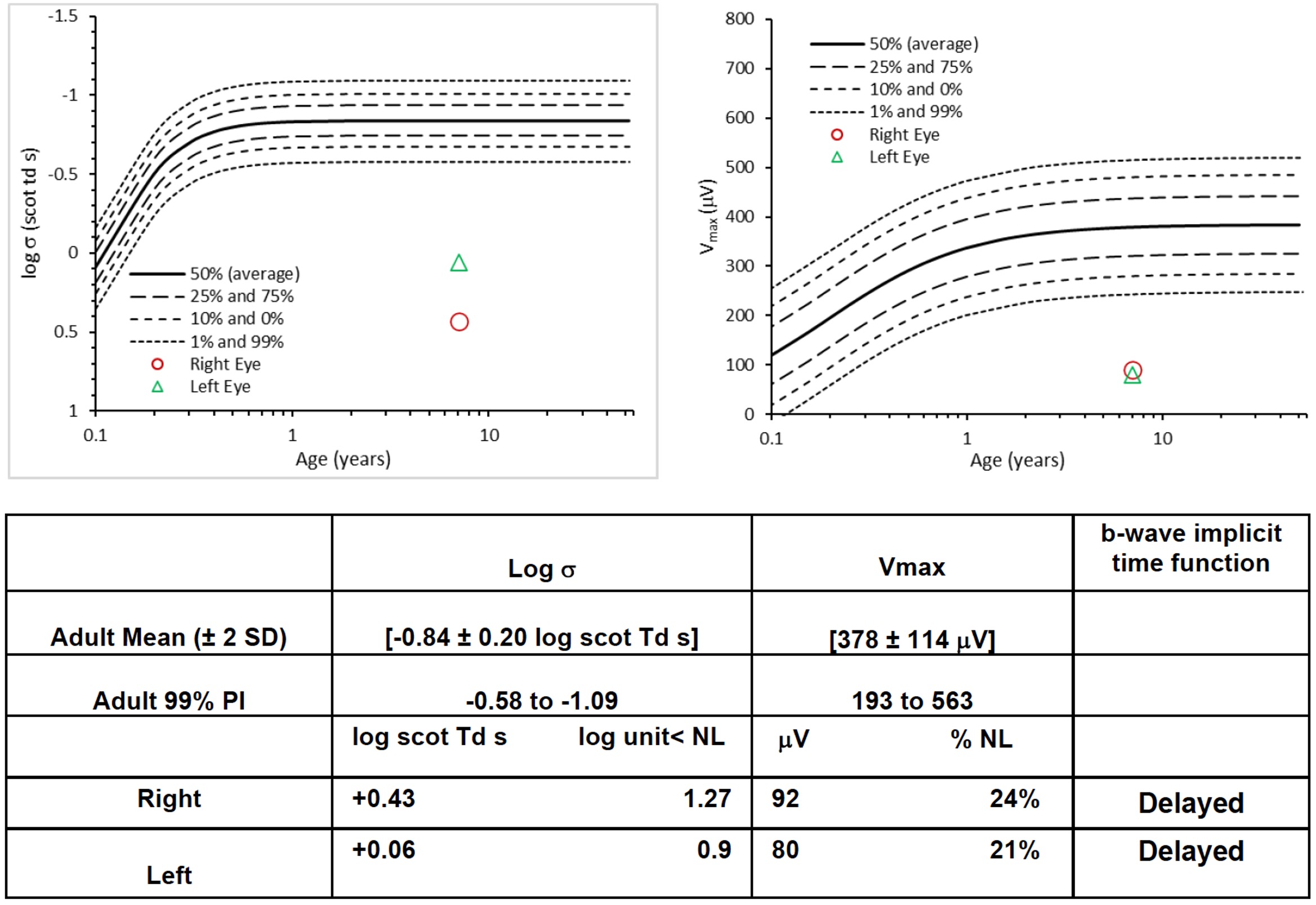
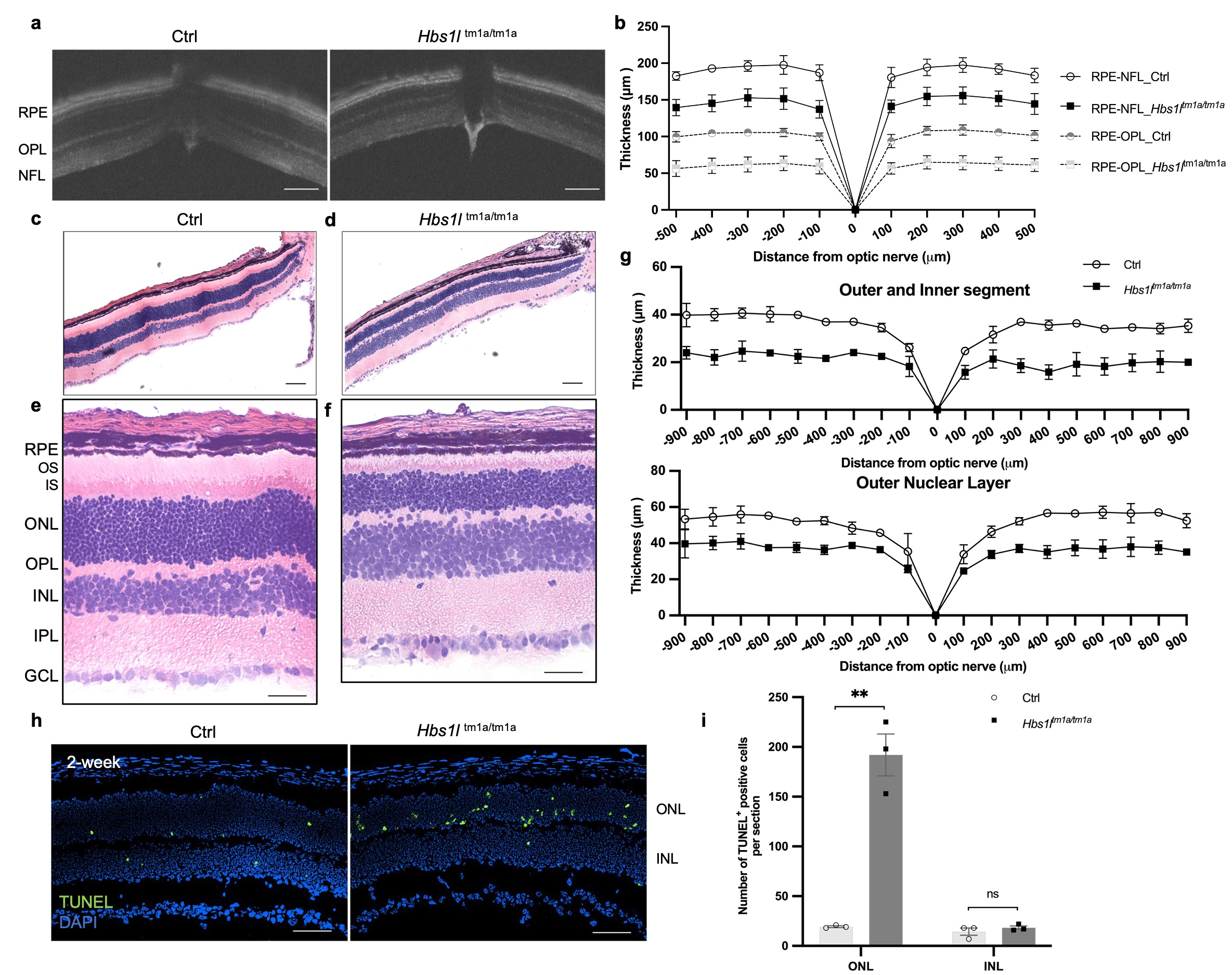
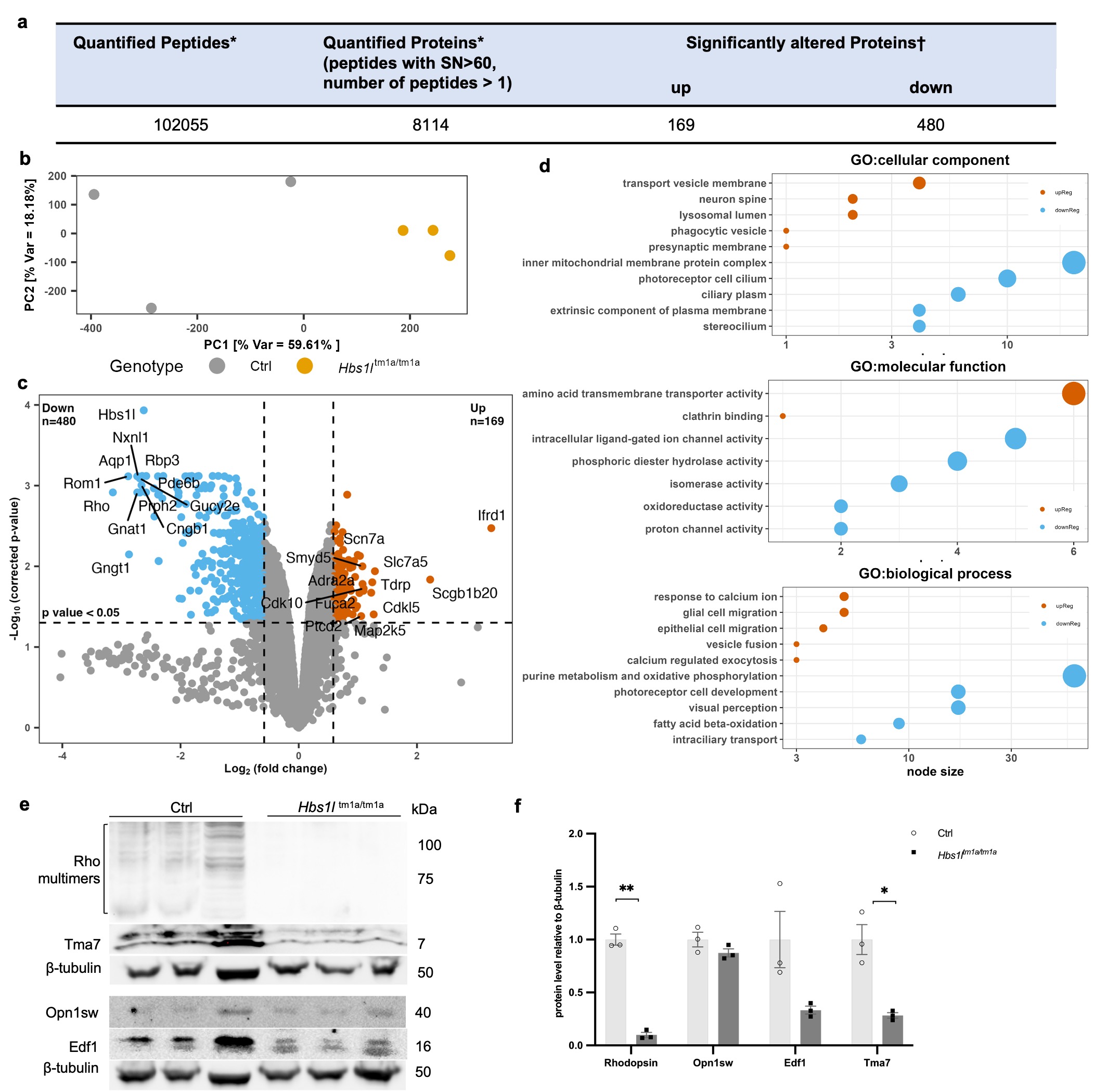
Background: Inherited retinal diseases (IRDs) encompass a genetically diverse group of conditions in which mutations in genes critical for retinal function are associated with progressive demise of photoreceptor cells and associated vision loss. Currently, over 260 IRDs-associated genes have been identified. While most mutated genes are expressed specifically in the retina, a few ribosome-related genes have been identified along with neurological manifestations. Objective: This study focuses on the HBS1L gene, a critical ribosomal rescue factor. Previously, we have reported a female child carrying biallelic HBS1L mutations, manifesting growth restriction, developmental delay, and hypotonia. In this study, we describe her ophthalmologic findings, compare them with the Hbs1ltm1a/tm1a hypomorph mouse model, and evaluate the underlying microscopic and molecular perturbations. Design/Methods: Bilateral fundus photography and electroretinogram (ERG) were performed to evaluate the retinal morphology and function in the patient. And in the Hbs1ltm1a/tm1a mice, a series of histopathological tests, in vivo imaging of optical coherence tomography (OCT), and an unbiased comprehensive proteomic profiling were performed to reveal the cellular and molecular changes following Hbs1l loss. Results: The patient was noted to have abnormal retinal pigmentation and impaired visual function, with dampened amplitudes of a- and b-waves in both rod- and cone-mediated responses. Hbs1ltm1a/tm1a mice exhibited profound retinal thinning of the entire retina, specifically of the outer retinal photoreceptor layer. TUNEL assay revealed retinal degeneration due to extensive photoreceptor cell apoptosis. Loss of HBS1L resulted in comprehensive proteomic alterations, with169 proteins increased and 480 proteins decreased including many critical IRD-related proteins. GO biological process and GSEA analyses reveal that these downregulated proteins are primarily involved in photoreceptor cell development, cilium assembly, and phototransduction. Furthermore, apart from the diminished level of PELO, HBS1L depletion was accompanied by reduction in translation machinery associated 7 homolog (Tma7), and Endothelial differentiation-related factor 1(Edf1) proteins, the latter of which coordinates cellular responses to ribosome collisions.
Conclusion(s): Genetic deficiency of HBS1L causes retinal degeneration in both the human patient and mouse model. This novel connection between HBS1L and EDF1 highlights the intricate mechanisms underpinning ribosomal rescue and quality control that are essential to maintain the homeostasis of key proteins of retinal health.