Associate Professor Johns Hopkins University School of Medicine Baltimore, Maryland, United States
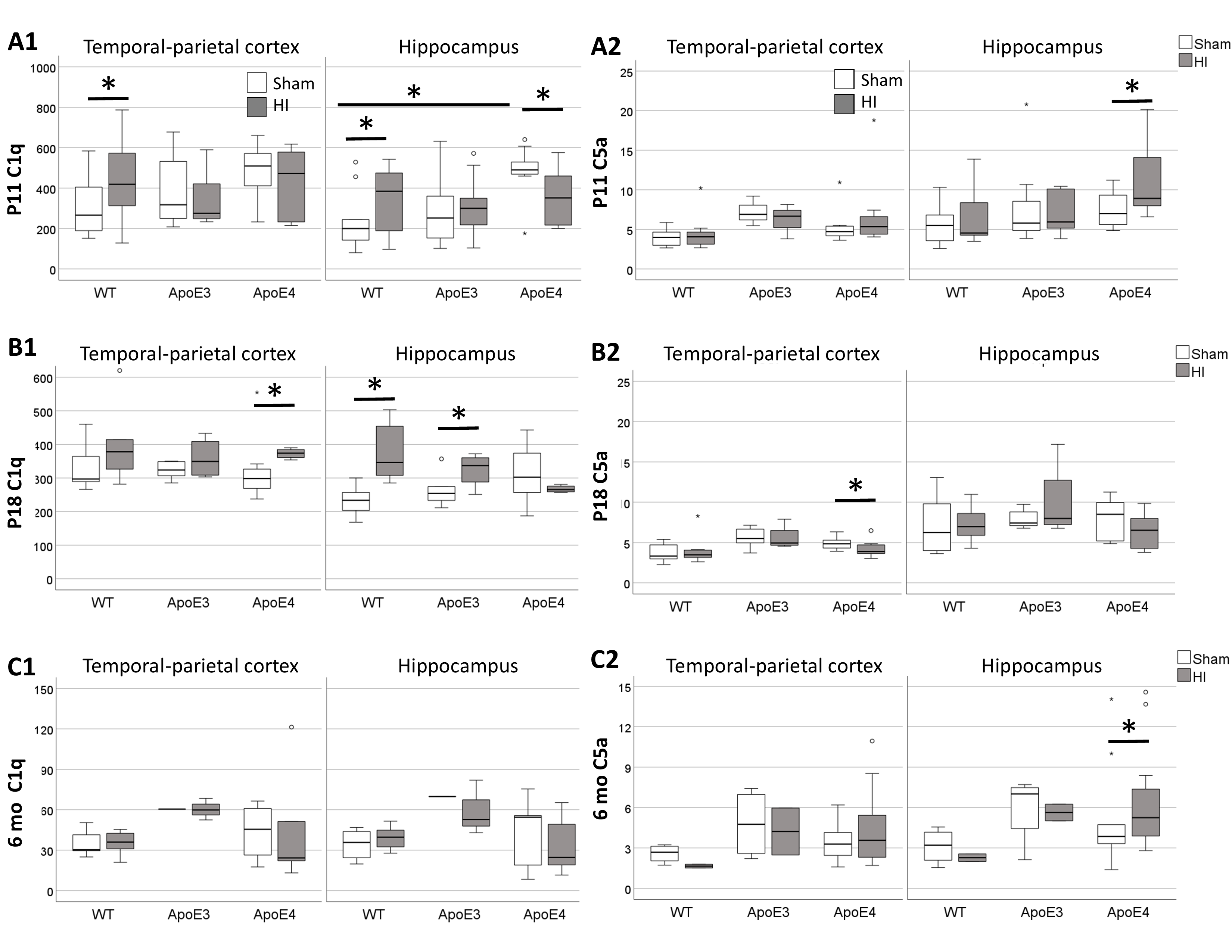
Background: C1q initiates the classical complement cascade (CCC) leading to early inflammation and later repair. C5a is a pro-inflammatory chemoattractant downstream from all complement pathways. ApoE is a known checkpoint limiting CCC activity via C1q binding. ApoE4 binds avidly to C1q forming complexes found in Alzheimer’s disease plaques leading to neurodegeneration. Objective: Hypoxic-Ischemic (HI) brain injury leads to C1q-mediated oxidative injury and C5a-mediated inflammation; thus, we aim to study if increased C1q in neonatal HI is modulated by ApoE4, resulting in greater C5a, injury and inflammation. Design/Methods: Cerebral HI injury (Vannucci model) was produced on postnatal day (P)10 ApoE humanized mice compared to wildtype (wt) C57BL6 mice and sham ApoE mice. At 24h, 8d and 6 mo after HI, we studied the parietal-temporal cortex (Ctx) and hippocampus (Hp) using a combination of immunoblotting and MSD for C1q & C5a (complement); IL-1β, IL-6, CXCL1 and TNF-α (pro-inflammatory cytokines) and α-fodrin breakdown (cell death marker). Results: At P11, C1q was >1.7-fold higher in HI-injured wt Ctx and Hp (A1), which correlated with increased cytokines (all p< 0.05) and 150kda α-fodrin fragments (r 0.59, p=0.006). Sham ApoE4 Hp had a 2.5-fold higher C1q than wt at P11, an increase prevented by HI. This relative deficit in HI-injured ApoE4 Hp correlated with higher 150kda (r -0.61, p=0.01) and 120kDa (r -0.53, p 0.02) α-fodrin fragments. We did not find differences in C5a 24h after HI injury, except for a 31% increase in the HI ApoE4 Hp (A2), which correlates with higher IL-1β and IL-6, but not markers of cell death. At P18, HI-injured wt and ApoE3 Hp had higher C1q (B1), which correlated with higher cytokines, but not cell death, with similar trends in Ctx. Although C1q increased in HI-injured ApoE4 Ctx vs. sham at P18, this only correlated with increased TNF-α (r 0.81, p< 0.001) and did not occur in the Hp (B2). At 6mo, only HI-injured ApoE4 Hp had higher C5a associated to increased cytokines (C2).
Conclusion(s): In sham ApoE4 mice, C1q upregulation may be a compensation from the ApoE-C1q inhibitory CCC checkpoint. In response to HI, C1q increase may be a leading acute mechanism of cell death and neuroinflammation in wt mice, while C1q decrease with C5a upregulation appear to serve a similar purpose via activation of the alternative or lectin complement pathway in ApoE4 mice. C1q has reparative functions at delayed stages of brain injury, thus depressed upregulation in HI-injured ApoE4 Hp, may lead to late C5a upregulation and neuroinflammation, as we have documented previously in the 6 mo mice.

 photo")